您好,登录后才能下订单哦!
密码登录
登录注册
点击 登录注册 即表示同意《亿速云用户服务条款》
这篇文章主要讲解了“qt MAF过滤的方法”,文中的讲解内容简单清晰,易于学习与理解,下面请大家跟着小编的思路慢慢深入,一起来研究和学习“qt MAF过滤的方法”吧!
经过去掉缺失,去掉错误的性别信息,得到的文件为:
HapMap_3_r3_6.bed HapMap_3_r3_6.fam HapMap_3_r3_6.log
HapMap_3_r3_6.bim HapMap_3_r3_6.hh
这里,我们根据最小等位基因频率(MAF)去筛选。
「为什么要根据MAF去筛选?」
❝最小等位基因频率怎么计算?比如一个位点有AA或者AT或者TT,那么就可以计算A的基因频率和T的基因频率,qA + qT = 1,这里谁比较小,谁就是最小等位基因频率,比如qA = 0.3, qT = 0.7, 那么这个位点的MAF为0.3. 之所以用这个过滤标准,是因为MAF如果非常小,比如低于0.02,那么意味着大部分位点都是相同的基因型,这些位点贡献的信息非常少,增加假阳性。更有甚者MAF为0,那就是所有位点只有一种基因型,这些位点没有贡献信息,放在计算中增加计算量,没有意义,所以要根据MAF进行过滤。
❞
「思路:」
「提取常染色体上的位点名称:」
因为这里是人的数据,所以染色体只需要去1~22的常染色体,提取它的家系ID和个体ID,后面用于提取。
awk '{ if($1 >=1 && $1 <= 22) print $2}' HapMap_3_r3_6.bim > snp_1_22.txt
wc -l snp_1_22.txt
1399306 snp_1_22.txt 常染色体上共有1399306位点。
这里,用到了位点提取参数--extract
plink --bfile HapMap_3_r3_6 --extract snp_1_22.txt --make-bed --out HapMap_3_r3_7
「查看日志:」
PLINK v1.90b6.5 64-bit (13 Sep 2018) www.cog-genomics.org/plink/1.9/
(C) 2005-2018 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to HapMap_3_r3_7.log.
Options in effect:
--bfile HapMap_3_r3_6
--extract snp_1_22.txt
--make-bed
--out HapMap_3_r3_7
515185 MB RAM detected; reserving 257592 MB for main workspace.
1431211 variants loaded from .bim file.
163 people (79 males, 84 females) loaded from .fam.
112 phenotype values loaded from .fam.
--extract: 1399306 variants remaining.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 112 founders and 51 nonfounders present.
Calculating allele frequencies... done.
Total genotyping rate is 0.998153.
1399306 variants and 163 people pass filters and QC.
Among remaining phenotypes, 56 are cases and 56 are controls. (51 phenotypes
are missing.)
--make-bed to HapMap_3_r3_7.bed + HapMap_3_r3_7.bim + HapMap_3_r3_7.fam ...
done.
可以看到,共有163个基因型,共有1399306个SNP
首先,通过参数--freq,计算每个SNP的MAF频率,通过直方图查看整体分布。可视化会更加直接。
plink --bfile HapMap_3_r3_7 --freq --out MAF_check
结果文件:MAF_check.frq预览:
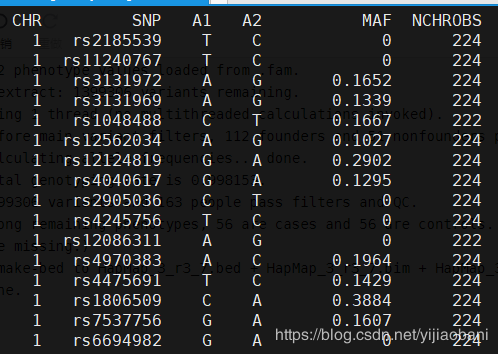
「R代码:」
maf_freq <- read.table("MAF_check.frq", header =TRUE, as.is=T)
pdf("MAF_distribution.pdf")
hist(maf_freq[,5],main = "MAF distribution", xlab = "MAF")
dev.off() 「图形如下:」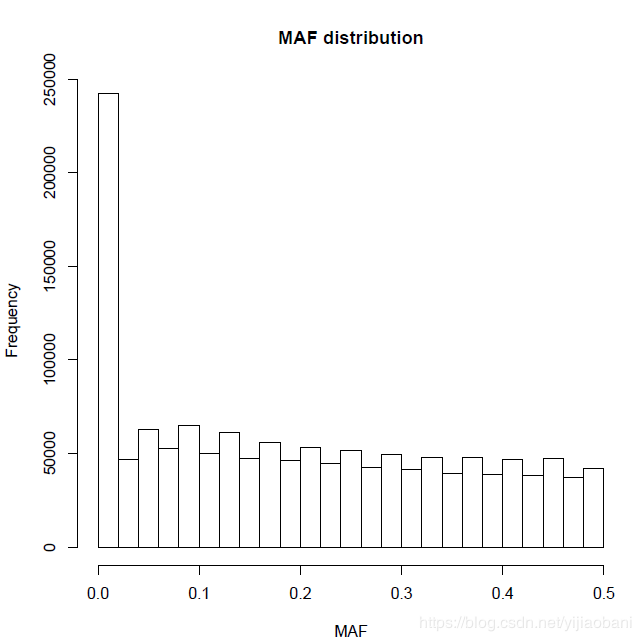
可以看出,很多基因频率为0,说明没有分型,这些位点需要删掉。
plink --bfile HapMap_3_r3_7 --maf 0.05 --make-bed --out HapMap_3_r3_8 日志:
PLINK v1.90b6.5 64-bit (13 Sep 2018) www.cog-genomics.org/plink/1.9/
(C) 2005-2018 Shaun Purcell, Christopher Chang GNU General Public License v3
Logging to HapMap_3_r3_8.log.
Options in effect:
--bfile HapMap_3_r3_7
--maf 0.05
--make-bed
--out HapMap_3_r3_8
515185 MB RAM detected; reserving 257592 MB for main workspace.
1399306 variants loaded from .bim file.
163 people (79 males, 84 females) loaded from .fam.
112 phenotype values loaded from .fam.
Using 1 thread (no multithreaded calculations invoked).
Before main variant filters, 112 founders and 51 nonfounders present.
Calculating allele frequencies... done.
Total genotyping rate is 0.998153.
325518 variants removed due to minor allele threshold(s)
(--maf/--max-maf/--mac/--max-mac).
1073788 variants and 163 people pass filters and QC.
Among remaining phenotypes, 56 are cases and 56 are controls. (51 phenotypes
are missing.)
--make-bed to HapMap_3_r3_8.bed + HapMap_3_r3_8.bim + HapMap_3_r3_8.fam ...
done.
可以看到,325518个位点被删掉了,剩余1073788 个位点。
「结果文件:」
HapMap_3_r3_8.bed HapMap_3_r3_8.bim HapMap_3_r3_8.fam HapMap_3_r3_8.log感谢各位的阅读,以上就是“qt MAF过滤的方法”的内容了,经过本文的学习后,相信大家对qt MAF过滤的方法这一问题有了更深刻的体会,具体使用情况还需要大家实践验证。这里是亿速云,小编将为大家推送更多相关知识点的文章,欢迎关注!
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。